| |
| |
| |
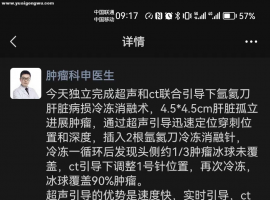 病例分享/肿瘤晚期全耐药,特殊治疗
病例分享/肿瘤晚期全耐药,特殊治疗寻生机
晚期肿瘤常规方案全无效了怎么办?
病例分享/肿瘤晚期全耐药,特殊治疗
病例分享/肿瘤晚期全耐药,特殊治疗寻生机
晚期肿瘤常规方案全无效了怎么办?
 请大神帮忙看一下81岁老人刚确诊肺腺
医生说不建议做手术,建议做一个基因检测然后吃药,但是医生具体的诊断结果也有没说,
请大神帮忙看一下81岁老人刚确诊肺腺
医生说不建议做手术,建议做一个基因检测然后吃药,但是医生具体的诊断结果也有没说,
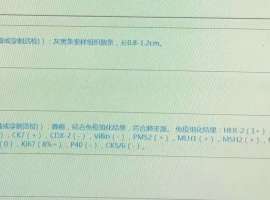
 肺腺癌晚期ⅣB,基因靶点KARS-G12C,
各位老师,我哥肺腺癌晚期,患有强直性脊柱炎,目前治疗方案为:贝伐单抗+化疗药,10
肺腺癌晚期ⅣB,基因靶点KARS-G12C,
各位老师,我哥肺腺癌晚期,患有强直性脊柱炎,目前治疗方案为:贝伐单抗+化疗药,10
 神奇的ommaya囊——学习笔记
一个神奇的装置-ommaya囊——————9.28 苗茜医生科普讲座
直播链接:https://live.
神奇的ommaya囊——学习笔记
一个神奇的装置-ommaya囊——————9.28 苗茜医生科普讲座
直播链接:https://live.
 求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC
求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC





 显身卡
显身卡






