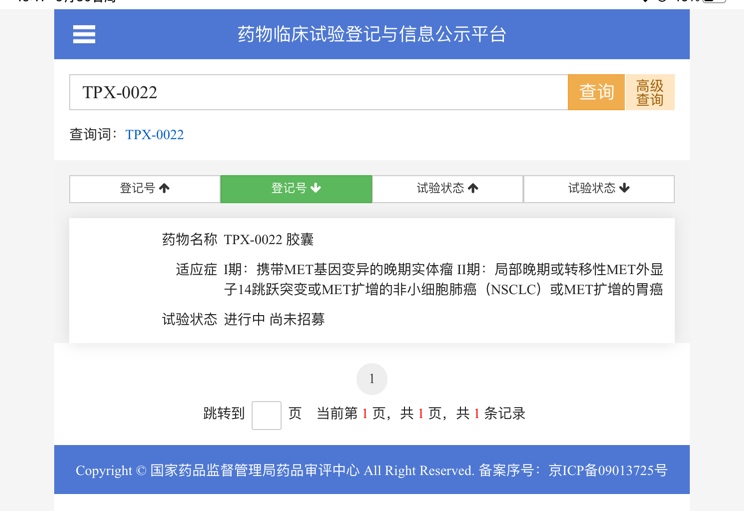
| |
 汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
 奥西伏美阿美耐药怎么办?基因全因没
你以为我想盲试?,这不也是没有办法的办法?谁不想正规治疗,按照指南和循证医学的节
奥西伏美阿美耐药怎么办?基因全因没
你以为我想盲试?,这不也是没有办法的办法?谁不想正规治疗,按照指南和循证医学的节
 塔拉妥单抗获批!小细胞肺癌治疗迎来
作者:seacat今年2月份举行的第23届中国肺癌高峰论坛主题是 “历史转折关头的小细胞肺
塔拉妥单抗获批!小细胞肺癌治疗迎来
作者:seacat今年2月份举行的第23届中国肺癌高峰论坛主题是 “历史转折关头的小细胞肺
 免费领取5斤洗衣液:掌握新药机会,
抗癌路上,信息就是力量。加入“与癌共舞”官方新药情报站,每天10分钟,就能掌握最
免费领取5斤洗衣液:掌握新药机会,
抗癌路上,信息就是力量。加入“与癌共舞”官方新药情报站,每天10分钟,就能掌握最
 70天肺癌转移灶全部消失,我打赢了这
讲述者:幸福快乐整理者:pear2023年4月,我被确诊为肺腺癌,那一刻,世界仿佛瞬间褪
70天肺癌转移灶全部消失,我打赢了这
讲述者:幸福快乐整理者:pear2023年4月,我被确诊为肺腺癌,那一刻,世界仿佛瞬间褪






 显身卡
显身卡