马上注册,结交更多好友,享用更多功能,让你轻松玩转社区。
您需要 登录 才可以下载或查看,没有账号?立即注册

x
肿瘤的靶向治疗一直受困于癌细胞的耐药性,由于癌细胞总是随机地产生基因突变,并由此导致异常的肿瘤异质性,这使得靶向药物的耐药是一个看似无解的问题。+ [2 b2 }; d8 @! l6 m
/ }! ?* j% r9 E t$ D- m: e" T目前对于肿瘤靶向药物耐药的解决方法是,在临床或者影像学进展后开始新的治疗(这里很多病友甚至在CEA等肿标上升、体感不适即更换治疗措施),但是最终导致所有的药物都没有用了。+ k ?: u8 G$ i9 \; u" H% |8 Y
1 V, y$ w: Y5 e: x% N
这篇发表在Scientific Report上的文章做出了一个替代解决方案,即通过计算机模拟来系统性地分析人类肿瘤的各种数据,这样可以很好地去识别肿瘤的动态情况,进而可以提前采取相应的措施,甚至有时采用一些与常规直觉相反的治疗措施,达到对肿瘤的实时的控制。癌度为大家编译和分享这篇最新文献,希望能给大家带来帮助。
" P H5 M5 ~& y( w, [2 ~/ a$ S6 S7 F7 m) y9 q
 : |9 e# X# G* Y
: |9 e# X# G* Y
1 O3 S1 j3 }# D( d# k- p靶向治疗是一个非常有力的武器,对于存在EGFR、ALK、ROS1、BRAF等肿瘤驱动基因突变的患者,使用靶向药物可以较好地获益,但是好景不常在,多数患者在几个月之内很快地耐药。
( w9 l; g5 W( F& t3 m
) C% ^& ^: ^5 ^" i肿瘤之所以耐药的原因就是肿瘤不断的随机产生突变,和基于这些突变所形成的肿瘤的异质性,这使得临床的治疗方案难以为继。因为很难找到一种靶向药物长时间地控制患者所有肿瘤病灶。靶向药物压制了敏感的癌细胞,但是耐药的癌细胞总是不断地、动态地产生,繁衍并促进了肿瘤的进展。
: P% @. P& }5 d. A, O- Z: l- e: Y% }) x7 R3 o) k
目前对于实体肿瘤的治疗方案都是静态的,除非是影像学或者临床明确进展,否则不会更换治疗措施。相反,设计一种动态的治疗策略,并实时地在靶向药物之间、或者靶向药物组合之间切换,将可能很好地抑制肿瘤的那些罕见的、突发的耐药癌细胞群,进而能达到对肿瘤的长期的控制,或者也达到了癌度所提倡、癌度船长正在实行的那种“与癌共舞”的生存策略。1 V3 Q" `8 ^( B" x& d& D! Y
6 K, H( ?3 @: O- }
说起来容易,但是该怎么做呢?, n& F; Y9 {1 a" M8 E: _" c& X/ W
7 a1 N9 h, N7 ]6 [1 ^5 Z
通过建立数学模型和计算机数值模拟,分析肿瘤生长速度、异质性和不同治疗方案的疗效,长期以来被用作洞察分析肿瘤进化模式和制定有效治疗策略的依据。这种建模包括使用随机或确定性的微分方程来描述静态或动态治疗肿瘤的各种癌症模型,而且使用这种数学模型还可以用来研究耐药性的问题。$ E4 s. m2 H' ?
B1 C, j3 H8 O4 }( C2 b( f$ b
 ( o- ^8 X) m, A& g" W' b3 F7 h0 x
( o- ^8 X) m, A& g" W' b3 F7 h0 x
) t; ?- ]4 {6 V% b9 G很多科学家对此兴趣浓厚,比如使用数学模型来研究淋巴瘤异质性的治疗方案。利用数学模型来研究肿瘤的最佳控制理论可以追溯到上世纪70年代,最近的例子则是抗血管生成治疗药物和化疗的联合、化疗和免疫治疗的联合。但是之前的数学模型大部分基于静态治疗策略,很少涉及动态治疗的模拟,而且缺乏对患者实践过程中产生问题的回应,如肿瘤的异质性、药物浓度的波动等。1 s1 N$ ?$ j8 v* ?% c3 R7 I$ ~
% e7 Q4 K$ B4 F1 s% l这篇文献的研究者设计了一种新的数学模型,这种模型结合了肿瘤细胞群的进化特点以及可检测到的一些肿瘤参数。
$ h/ I# }% s8 Z+ }% X# @. r* o1 Z7 V) j
研究的问题主要是下面三个:
9 E1 \2 H4 {9 [) h( e/ D
, U3 _3 {7 ^5 Q/ D/ H肿瘤的遗传组成和降低药物剂量如何影响治疗方案的疗效和有效时间?
, f r" _- d" i# [2 r8 ~9 v5 V F" y6 i" J8 z) O
对于小分子靶向药物的联合治疗,使用怎样的最佳剂量组合,可以更好地解决肿瘤的异质性、肿瘤的基因组进化和调控药物剂量波动?/ g' v0 Y9 U$ }4 F
5 ]% M* m! t/ {2 ?
对肿瘤患者进行连续活检,或者是基于ctDNA的连续液体活检是必要的。
. F& D& x* J) ~0 V
% P7 c* |! C7 g但什么时候是最佳时机? u5 l& f& D; }! N, C0 {
2 ^% y$ ]+ W6 J6 m
研究者提出了几个新见解:/ `9 T/ _# q X+ S( f6 B
7 R! V @9 [: g1 ]0 r1. H4 ]" t$ q3 F8 F* h; M
多种治疗措施的动态切换(主动轮换)可以很好地控制多个肿瘤细胞亚群,也就是控制肿瘤的异质性。静态的双药组合不能有效地控制所有的肿瘤细胞亚群,即动态地调整比静态的要好。
0 p. ~9 v4 T2 m) i7 a" E: J" ~, a( |" E
2- Y$ l4 L. t$ ]! ^/ y
对于异质性很强的肿瘤细胞群落,恒定的药物组合在药物浓度方面是缺少变化和波动的,更可能导致肿瘤快速进展。通俗的语言就是,药物组合不怎么变化,癌细胞面对的药物浓度也是稳定的,时间一长就可能适应药物产生耐药。
b- V. t. g2 u
" h! H3 N: u" S* X x0 l4 ^2 E3' { R( p- r9 [# K) F/ G
在肿瘤病灶对治疗措施响应的时候,主动地在多种治疗措施中进行切换,可以有效地阻止耐药性癌细胞亚群的生长,这就是说在临床症状或影像学指标变化出现之前,就把更早出现的肿瘤耐药分子层面的进展给处理掉。这是很难抉择的,尤其是在肿瘤对治疗应答很好的情况下。癌度带大家来看看相关的案例。
' R' C$ O$ @; ]' @- Y. e, H) { s+ ^, h% X& h+ ~
8 t8 a0 \' o' REGFR突变的肺腺癌患者的肿瘤遗传异质性、以及这种异质性的进化( P* W2 d$ F, S7 i
, w! Q4 v6 K9 H* w由于肺腺癌的靶向治疗、耐药机制研究的比较清楚,而且单个肺腺癌患者就存在复杂的肿瘤异质性,靶向药物的耐药原因有可能是涉及多个方面。因此克服这种多基因的耐药策略极为重要。
# J# V3 a6 i U. a$ @2 L) A O( p- R x! w, [7 d* H0 x+ K
 7 ~! s; E3 E6 V g2 o7 |
7 ~! s; E3 E6 V g2 o7 |
* N9 p0 m) V) j$ j& l1 e
研究者为了验证他们的计算模型和理论,以一名41岁的肺腺癌患者为研究对象,这名男性患者没有吸烟史,存在EGFR基因的21外显子L858R突变,但是患者在使用厄洛替尼(特罗凯)四个月后就耐药了,远低于EGFR敏感突变的肺腺癌患者的9-12个月的PFS。
) r4 q, A2 d. x5 T) y0 J) } u. g( Z: L5 V) J/ Y
研究者通过二代基因检测技术,对389个肿瘤相关基因的外显子和部分内含子进行测序,使用的组织样本包含特罗凯治疗前和耐药后的样本。在治疗前的肿瘤组织里发现了BRAF基因的V600E突变(突变频率为6%),有报道该基因突变(RAF-MEK-ERK通路激活)会导致EGFR靶向药物耐药。在特罗凯耐药后的检测样本中发现BRAF基因的V600E突变频率增加了10倍。这是因为携带BRAF V600E的癌细胞数目增加了,另外研究者还发现MET基因也发生了扩增,还有一个低频率的T790M突变。! R" \1 M5 Z; [7 V5 p, d# ]
; D" U) Y5 ]# U2 B7 e2 x: s

' Q. N8 E6 a( P8 n( E9 J
* {8 x L8 q* R2 E6 u+ C4 [6 K' M+ T0 D图1:A:患者使用特罗凯6周后肿瘤基本消失,4个月后复发。B图的浅色柱状图表示治疗前的肿瘤,深色的柱状图表示耐药的肿瘤,可以看出特罗凯治疗前BRAF基因V600E突变就存在,在耐药后频率增加了十倍,而EGFR基因的T790M是耐药后产生的。1 E7 g% T! q9 L/ o2 a" d0 b8 x5 f
3 M9 S0 c& w* Z4 x, v0 e) {

- Q; _" |1 S* C* j0 O; n; D/ s
) h: e7 i# d* F! w" w/ `图2:治疗前后DNA拷贝数的变化情况。特罗凯治疗前EGFR基因的21外显子的L858R区域的拷贝数很多(浅蓝色圆点)。在特罗凯的选择压力下,EGFR基因的21外显子的L858R区域的DNA拷贝数下降,而MET、BRAF基因的拷贝数开始上升(深色蓝圆点)。
7 {$ C% ?/ ^" ?% \/ d
' @* Y3 Y9 x9 m& W由图1和图2我们可以看出,该患者的特罗凯耐药涉及到BRAF基因的V600E突变、EGFR基因的T790M突变和MET基因的扩增,是一个涉及到多重机制的耐药情况,最终导致了患者对特罗凯快速耐药。
% F. l0 B9 j) {, E. t( t) I) V' R
细胞学实验表明:
5 T e4 _+ A+ U8 @, K' U
! f9 d7 t, t2 P8 N1 DBRAF基因V600E突变的拷贝数扩增导致了EGFR的L858R对特罗凯耐药,而野生型的BRAF基因不会产生耐药。
: m# K4 _9 f; ?0 y, j2 L# T
5 n" A |7 z/ }对于BRAF基因V600E导致的耐药癌细胞,使用特罗凯联合威罗菲尼(BRAF抑制剂)或者曲美替尼(MEK抑制剂),可以解决耐药的问题。
, ] H7 S0 Z/ Z, A
1 A! ^% @5 J" E+ j如果在这个癌细胞模型里再使用肝细胞生长因子(HGF)来激活MET,即模拟EGFR靶向治疗时的MET扩增,结果发现MET的激活不仅导致了特罗凯的耐药,还同时对BRAF基因V600E的表达具有促进作用,这主要是通过增加MEK、ERK和AKT的磷酸化来实现的。这种情况下使用BRAF抑制剂威罗菲尼,或者MET的抑制剂克唑替尼都不管用,而使用MEK抑制剂曲美替尼却可以解决癌细胞模型的这一耐药问题。
. ?- J; g( @+ X- w g) ?# C$ ~9 j8 `) z& t5 ]( N; }
对于EGFR基因T790M、BRAF基因V600E和EGFR基因的L858R的关系,研究者在H1975细胞系进行了研究(该细胞系携带EGFR的L858R、T790M),研究者发现BRAF基因V600E可适度降低H1975癌细胞系对阿法替尼的敏感度,当然这个降低可以通过联合威罗菲尼这个药物来克服。& B5 [- B: u2 I, i5 G+ R g- C8 c ?6 o
' I/ q9 V$ _$ x- ]! U- x3 N特罗凯治疗导致了癌细胞产生了多个基因突变,这使得肿瘤的遗传背景极为复杂,解决肿瘤的耐药问题需要EGFR和MAPK信号通路的多个药物联合才行。
1 ^; I3 |3 z1 o( ~$ B' p1 H# j. Q5 Q: T/ O% _1 x8 V" T
利用一种特殊的癌细胞系,研究者模拟了这名患者特罗凯耐药后肿瘤细胞群的遗传背景,通过不同的药物组合、更换治疗药物的时间最终得出一个最佳的策略。如下图所示:* z1 i- t- s% C
7 L5 R5 E6 g, V( A5 t- m
 ) N5 k2 S/ ?/ w; ^
) N5 k2 S/ ?/ w; ^
+ f! G. Q1 j' L/ `5 J4 Z7 f
图3:动态轮换靶向药物组合,更好的抑制癌
F" |$ q$ L( |2 O$ N5 m5 w) @! X# s1 g- ]- a& c5 F9 |4 o; ^5 ?
细胞亚群
- w% |2 w! \8 U1 o% F3 e. G: A3 p; A/ F3 Q
如图3所示,研究者使用了一种癌细胞群,与上面的那个41岁的肺腺癌患者的情况类似的遗传背景,这个癌细胞群有三种癌细胞,它们分别是:$ n6 C5 f3 }. N
`7 x% {& n/ n3 {
A群癌细胞(占比89%):只携带EGFR基因的21外显子L858R突变,占比89%,对厄洛替尼、阿法替尼敏感。
( m6 O" L# o+ ~$ M$ \0 m* G9 Y: D7 ^$ X
B群癌细胞(占比10%):同时携带两种突变,EGFR的L858R突变,和BRAF基因的V600E突变,对于厄洛替尼联合威罗菲尼或者曲美替尼敏感。
) ?! R' G# Y- D9 n/ Z! f5 ?6 G7 e n/ o/ g
C群癌细胞(占比1%):同时携带三种突变,EGFR基因的L858R,EGFR基因的T790M,以及MET基因扩增(这里使用HGF来模拟的)。在上面我们介绍过这种癌细胞群应该使用阿法替尼和曲美替尼。这里为何不是用AZD9291而是用阿法替尼解决T790M的问题,是研究者的选择,后续再讨论。
" ?7 k! d( Z+ B: H/ B R% R o, i2 z5 A$ g0 @& w$ {
研究者分别使用两种药物组合轮换,或者一直用某两种药物,来抑制最初的癌细胞群,我们来看看这四种治疗方案的效果如何。
0 Q3 o' Y: [4 n0 m( [6 d& {+ M( h+ E, q" _
1
; o; n# a/ \2 S5 U+ v$ O# G计算机模拟的最佳治疗方案是:
0 j: q2 H2 K2 h" r9 j& D
- w. I( T4 K* C' F6 M9 G7 N1-5天使用厄洛替尼联合克唑替尼,6-30天使用阿法替尼联合曲美替尼。) j& {3 s t; G( Y/ Q r
8 d8 J2 N& P5 l癌细胞总数呈现出持续下降,下降幅度为100倍(10^5到10^3),计算模拟显示这种治疗措施,有一条漏网之鱼,那就是携带BRAF基因、MET扩增(HGF+)的癌细胞,上图中的紫色曲线,不过即便是这个复杂的癌细胞群,也会被逐渐地抑制。
* _- X% |" t' n
) P6 j% b, b7 Z% @+ z- d9 ~6 l2
) M1 c& ~/ Z" W4 r% x+ \与方案1用药一样,只是更换的时间不同
" J/ r0 i* @; n6 b& L
) B0 d- S1 {# l( e7 t' [8 k1-10天使用厄洛替尼联合克唑替尼,10-30天使用阿法替尼联合曲美替尼,也就是只是比最优方案换药晚了5天而已。
9 ?+ W# f1 Y: E2 i, m0 O6 ~' ^# y9 E% i. l N6 I
这个效果就明显差一些,表现在癌细胞总数在治疗第10天有个抬头,达到了最初癌细胞数目的10倍,而且最后更换至阿法替尼联合曲美替尼后,癌细胞总数为10的4次方,比最优方案1的最终癌细胞数目多了10倍。而且中间图的紫色曲线不再表现出下降和被抑制的趋势,而是一直平稳。
8 ^8 ?7 r+ s- ^$ ?) i# p
# F0 G5 \$ H! ]3
! r# U0 Y2 J2 n- @7 l' |4 d再来看连续用药的情况:
; @8 [3 `$ |4 x) B7 F# \6 \5 b9 `; z" E% n
即1-30天,一直使用阿法替尼联合曲美替尼。! t1 j% k2 m" l3 K, F; s& V0 Q- f
* Q6 f/ I! [2 X% j7 D8 \! l
这种情况下癌细胞总数倒是一直呈现下降的趋势,不过30天的时候癌细胞的总数为10的4次方,不如最佳治疗方案。计算机模拟显示这种用药方案不能很好地抑制同时携带EGFR基因L858R和MET扩增的癌细胞(蓝色曲线)。与紫色曲线不同,蓝色曲线是很可能会出现的,因为紫色曲线是同时携带BRAF基因V600E和MET扩增,这种情况下是癌细胞丢失最初的EGFR驱动基因,这几乎不可能发生。
9 c8 s8 Q& Z+ W1 W% }: @* `8 ?6 N* G9 u W- O
另外有意思的是,为何阿法替尼联合曲美替尼这种药物组合一直有效,但是对癌细胞的抑制情况不如方案1呢,尤其是方案1还表现出换药时一种癌细胞总数的上扬,不过后来的下降幅度就大很多了,可能是厄洛替尼联合克唑替尼治疗5天后,耐药的癌细胞,即同时携带EGFR基因L858R、T790M和MET扩增的癌细胞迅速增加,在更换至阿法替尼联合曲美替尼时,这部分癌细胞被快速打压去了,导致癌细胞总数目的迅速下降。 Q* t# O" @! n1 n7 a5 I
9 W# q& P8 X% E/ {0 P3 x* ~
4
& t* x/ E* R# Z9 p- _+ ?最糟糕的的情况是
. G( E$ F7 o5 j/ \8 D: x
/ \0 s1 j6 x! i/ d( T. N一直使用厄洛替尼联合克唑替尼治疗。* @! x8 t0 S" q! @6 R f
, }2 S [$ ~3 ?+ M I5 O9 e: C5 f除了最开始有效之外,很快就耐药了,同时携带EGFR基因L858R、T790M和MET扩增的癌细胞迅速增加(褐色曲线),这使得癌细胞总数快速增加,30天后到了10的12次方。% s9 @! F7 E* O: F
* Y2 ~ h) S- e5 o5 R# k' T3 \
通过下面这张图再次加深认识一下四种治疗措施的效果。
5 s* q# a8 B @$ s) R3 g& `0 M& e G

! P# m& s2 g1 U b! r; S: j( u$ ~* L
图4:更换治疗措施,可以提高对肿瘤细胞群的综合压制效果
3 |$ X* {% h% H6 v3 K6 S
& y5 @6 H: U- ^/ o) |$ n6 @! m! _如图4所示,厄洛替尼联合克唑替尼治疗5天后,更换至阿法替尼联合曲美替尼治疗(6-30天)可以达到最佳治疗效果,上面一排中间的培养皿里癌细胞的总数最少的。当然最糟糕的情况就是厄洛替尼联合克唑替尼治疗,几乎癌细胞都长满了。
' F& x0 s& r4 X# Q' f
/ u2 y+ B$ K4 _6 [- w4 y这是一篇非常复杂的文献,癌度也并没有尝试将所有内容编译完成,理论和机制性的东西看起来既枯燥也不易懂。# v- C" V7 b! V% F8 I. o i
2 ~' X$ C& Z9 e' R' G
文章的中心意思总结起来就是:
3 x$ x+ N0 x' I& x
. w( i# X$ I# L; P* r以一个EGFR基因L858R突变的肺腺癌患者的耐药情况来说明耐药机制的多种可能性和复杂性,会涉及到多个肿瘤基因突变的同时发生,形成了一个复杂的癌细胞群落。7 F5 M- V% d! q5 O0 E; V! @
4 |4 l' s7 a% X8 y' U! g
基于对这名患者特罗凯(厄洛替尼)的耐药后的基因突变情况,利用体外细胞试验和数学模拟的方式,测试两种靶向药物联合,以及多种靶向药物的轮换,寻找到了一种最佳的轮换策略。
' v/ N& ^+ k/ \( X7 f) l/ ^& ?, ?; i+ ?' C' ~. ~
最佳药物组合、轮换策略不仅仅是药物的种类,还有时间、时机的问题,同样的药物,一种情况5天就换可以达到最佳抑制效果,另外一种是10天后再换,对癌细胞的抑制效果却会弱不少。但两种方式,都比持续地使用药物,不进行轮换的效果要好。
( f6 s# R7 F) m/ W( q* W% P4 k0 o: R) y4 D, _
我们读东西,贵在获得一个认知,以更好地指导我们使用药物,抗击小癌。
) x: B: Q3 p% r9 D o
( _$ O6 i5 t: E0 U# _翱宇博士曾经看过一个会议直播,一个大牛讲我们在思考肿瘤耐药的结果时,也就是要想了解最终是什么导致了耐药,一定要结合现在所使用的治疗措施,如特罗凯治疗多数导致T790M,而奥西替尼(AZD9291)治疗后,多数会逼迫出C797S突变。当然这并不绝对是这样,如本篇文章讲的是BRAF、MET和T790M多个耐药突变几乎都在快速地产生了,导致了耐药的复杂性。
! a8 c% a! q& h2 N1 E2 z, i0 y* J+ |! H. k+ a1 ^5 G3 q
本篇文章的思路和理念无疑是有其合理性的,即在临床症状、影像学指标改变出现前,就在分子层面处理掉耐药的癌细胞。2 \" O! G; P5 I& ]1 F
0 z, ?; O) T9 P
 & U! M8 C! k! X* N- X
& U! M8 C! k! X* N- X
/ x3 I: y. f% h J怪鱼博士也根据临床试验和病友实践结果,总结过靶向主动轮换的个人思考:
5 w, Z) T& G9 J& C; J. U2 e Y- G% O* j# L8 ^/ T3 H( e7 ?- M
17 j/ K" A9 X1 K `* _
主动轮换的策略不能盲目效仿,不单纯需要二代测序等新的技术,更需要患者和家属对基因通路、医学、药物学有一定的了解和较强的学习能力,同时对体感有很灵敏的感受,还要辅以良好的身体和免疫机能。
4 y) E6 o, L/ }1 \
) S$ `! Y4 g) v" d! g# G2 [1 k; {7 I8 b
同一个通路的上下游靶点的轮换打击,一般很难取得效果;不同通路的打击有可能产生协同效应;驱动基因和非驱动基因的联合或者轮换打击有可能取得良好的效果。
- b1 R7 \0 E9 ?& n* J
+ Y# n% ?! w" c7 f2 f& V3
# y: _) Y% }: V& ]& R& ~同一靶点的不同药物,一种耐药了,另一种也可能有效。但前提是:后一种药物具有更低的半抑制浓度(IC50)、更稳定的血药浓度分布、更强的药物渗透率、更长的半衰期、药物与靶点的结合机制不同等因素。
" K& {; g' T9 T* _! g0 M# [. r
关键问题是( r$ N% H, T/ @4 L/ {& Y) p
2 ~9 Q; G2 @& H- e/ H/ K我们怎么知道哪些耐药的癌细胞已经产生了?
! W; t) k# y" l$ r
3 M9 o& d# C+ U: z2 e- z+ j它们是携带怎样的耐药基因突变?
, f% u9 j- @" D3 b5 ^
^& Q' f2 B" _. ^1 ~! h它们的频率是多少?/ U& _ ]+ u D1 g. x" Q
% f' ^! a6 K+ b我们是否能找到合适的靶向药物组合去压制他们的部分癌细胞,什么时候轮换到另外一种靶向药物组合合适?( g) ], Q; Q |
7 ^- o5 b% g9 W这篇文献表明要实现药物的疗效,一般都需要超过临床剂量,那么最佳的剂量是多少呢?
4 r7 U( {7 _+ {5 ~0 b2 `; _
- U2 q5 T0 H8 E: h0 r" m
 * ^$ m4 |- {; T0 Y# T
* ^$ m4 |- {; T0 Y# T
0 T4 F% U$ L2 v/ l
很遗憾,这些问题很多没有特别准确的答案,但是总体而言,癌度为您提供一些建议供您参考,或者是我们的一些思考。
! w9 h/ X8 n" X/ L- _$ g9 u! C- U7 P4 ?2 B9 E" L: M6 H2 t
1. 如果我们能及时地、准确地监控到肿瘤的遗传性改变情况,也就是有没有新的突变产生、突变产生的频率是多少,突变的基因有没有药物?是否检测方法完全准确无误?这样我们就能很好地控制肿瘤,即使没有药物,我们也可以使用其他传统治疗措施去适当地牵制。所以第一次做基因检测,一定要尽量使用组织多测序些基因,不要漏检,导致一些原发性耐药突变漏检,耽误治疗病情。+ E( f2 S+ I2 H) `
% g' F2 h. `, y) b
2. 如果要去实时地、全面和准确地了解患者的肿瘤基因突变情况,目前来说这还是不现实的,因为肿瘤的异质性,以及现有检测技术指标的灵敏度仍不完善,我们可以期待的是外泌体、表观遗传学等新的检测策略。目前的检测技术、价格、灵敏度和特异性还不足以让人满意,最主要的是监测的时机仍然不够明晰。
: ]. _7 i {# d7 z$ F- B! _
/ _( D' W4 A, ~- W3. 要根据患者现在使用的靶向药物,可能的耐药方向,来选择性地准备对应的药物,由于目前没有特别好的办法,能准确有效地检测到耐药细胞的分子改变,所以什么时候换药,怎么组合需要去摸索,当然我们希望患者和家属在条件许可的情况下,可以做下检测再去判断。
( M( w4 L$ ~. K- l7 ^( O' [1 X+ |+ S. r+ a# \. N, P8 d2 v' L
4. 如果肿瘤细胞是动态的,进化的,那么固定地使用一种治疗措施直至耐药,这可能不是最佳策略,如上文所述厄洛替尼的选择压下,不是只产生一种常见的T790M,这样就只使用9291即可,而是多种耐药突变同时发生,很多时候这种耐药突变是相互独立分子事件,最好的策略是及早地发现他们并清除。7 N4 {' V3 R9 C- u5 ?# ?
8 a% r# x7 ?: j) Q5. 这种治疗举措是比较新颖和前沿的,您可以参考和学习,但不建议盲目尝试。天下没有完全一样的癌症患者,所以任何患者耐药后的癌细胞遗传突变、癌细胞之间的比例关系都与本文的案例可能不同,这也决定了您不能直接拿本文的最佳治疗方案来试验,不管是药物组合还是换药时间都是如此,我们学习的是别人的思维方式,而不是完全的照搬。) M. F9 S3 o% P$ H* k3 u1 M5 d1 p
+ V. i% m/ G. Y) W* I! E% |

5 ?4 V3 w, l( r- |% j9 P( O% T( q0 K- x7 Z1 ?
在我们编译这篇文献的时候,惊悉“憨叔”(憨豆精神,靶向轮换民间实践第一人,病友们亲切而又尊敬的称呼他为“憨叔”)离开了大家,癌度全体同仁和大家一样心情十分难过。
& \3 C" _7 x- y, p6 E- I
4 e2 ^ W0 V( M% B: B憨豆先生以神农尝百草式的以身试药开辟了“靶药轮换”抗癌方法的实践,并总结出经验教训,写下一篇篇心得体会,诫示后人。更答复了病友们上万条咨询,传播抗癌经验帮助他们走出癌症阴霾。
1 S! N" {' n* s9 C4 ]: Z; y
% v: e9 d& o, q3 v& h' M8 L$ T3 H在伟大的憨豆精神的指引下,很多病友在生命被判只有短短几个月时,没有放弃,没有妥协,他们选择积极勇敢面对,更加去热爱生活、享受生命的每一分钟。4 z' X" Q# p2 Q
; A+ Y7 a9 x; N6 H: A& v在治疗中,他们不断学习、冷静思考、大胆尝试和综合治疗,延长了生命,创造了奇迹。同时,以知识经验爱心不断帮助中国几百万的癌友。憨豆精神虽然走了,但他的精神永存,希望永存!# S9 E% ~3 t+ X1 c: K5 E$ t
# R8 m4 c' b' x世界上的我们是同一趟车上的人,只是有些人早点下车,有些人迟点下车。无所谓先后,终究都要下车。生老病死,是大自然的规律。我们能做到的是在有限的时间里活的更精彩。死亡终结了生命,但没有终结感情的联系。7 O, Z# e( W: G5 |# @( H* a8 r
, C' p6 e4 T- X- |谨以此文向憨叔致敬!愿憨豆精神永存!群体抗癌,与癌共舞~
r2 U; L9 e/ }% S4 F3 o; J/ d0 _+ L' A& F) B$ }
参考文献:Jonsson VD, et al., Sci Rep. 2017 Mar 13;7:442063 P1 e* a+ p4 x4 ?: {/ t
4 t+ i+ U. A) x8 V
癌度已经细分出肺部肿瘤群、小细胞肺癌群、妇科肿瘤群、胃肠肿瘤群、肝胆肿瘤群以及小癌种群,并在各群配备专业人士及癌度大神,帮助大家解决治疗过程中的各种问题。) S$ {* P6 `- b* q# u8 K# Z2 T
* e) o7 e3 [3 X, o- \- U5 S有需要的朋友,请加微信号:17191208048或17191096158,癌度家人等你哦~6 A6 n0 v2 x! f2 [2 x
! X! ]$ G. o; c% @% X

: z7 [ M6 U5 R+ D

|
 都说免疫不入脑,入了就不得了
免疫治疗以后的疗效反应,当病灶增大时,不一定是进展,还有一种可能,叫做CR,这个现
都说免疫不入脑,入了就不得了
免疫治疗以后的疗效反应,当病灶增大时,不一定是进展,还有一种可能,叫做CR,这个现
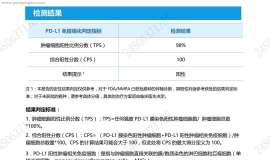
 母亲的基因检测和PD-L1报告出来了,
看了基因检测报告,似乎没有合适的靶向药,母亲确诊肺腺癌晚期,求助大家帮忙分析一下
母亲的基因检测和PD-L1报告出来了,
看了基因检测报告,似乎没有合适的靶向药,母亲确诊肺腺癌晚期,求助大家帮忙分析一下
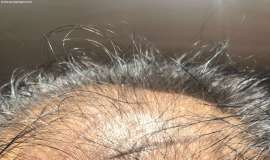
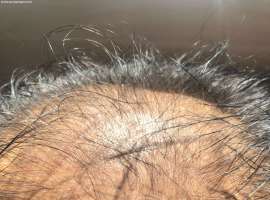 头部长个包 免疫性副反应吗
我们已经治疗将近四年了 一直用的单免疫治疗 没有用其他治疗 之前用的信迪利 今年初
头部长个包 免疫性副反应吗
我们已经治疗将近四年了 一直用的单免疫治疗 没有用其他治疗 之前用的信迪利 今年初
 晚期肺癌经过4次换药,如今母亲已经4
作者:pears
“万事开头难”这句话在母亲的抗癌路上体现得淋漓尽致,最初确诊肺癌后半
晚期肺癌经过4次换药,如今母亲已经4
作者:pears
“万事开头难”这句话在母亲的抗癌路上体现得淋漓尽致,最初确诊肺癌后半
 既要,又要,还要,什么药品可以治疗
作者:seacat
EGFR突变可以分为常见突变和罕见突变。常见突变就是EGFR外显子19del突变
既要,又要,还要,什么药品可以治疗
作者:seacat
EGFR突变可以分为常见突变和罕见突变。常见突变就是EGFR外显子19del突变


 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡